Assembly vs. Alignment
I promised in my first blog entry to explain what I'm interested in and why. I mean, we sequenced the human genome a decade ago (An Overview of the Human Genome Project), right? Why are we even discussing this?
The first relevant fact to note is that there are really two operations in which bioinformaticians are interested: assembly and alignment, but what do these terms really mean? In order to answer that question we need some background on the process that the actual DNA is put through and before that we have to have some info on genetics.
Genetics 101

 Step 0
Step 0
Before anything really fun happens, the DNA strand is cut up into little pieces for reasons explained below and injected into bacteria which are then allowed to reproduce, thus giving us many copies of each piece.
Step 1
The first step to the process happens on the chemical/physical level. The has to be run through a machine (called a sequencer) that reads each and every base pair but we do not have the technology to read all of the base pairs at once so we chop the DNA strand into smaller pieces. How long these pieces are is dependent on the sequencing machine and technology being used, but for our purposes there are two types: long read and short read. Long read sequencers work with strands several thousand base pairs long, whereas short read sequencers give data only a few hundred bp (base pairs) long. Long read is more reliable and easier to assemble but it is much slower and more expensive. Short read sequencers can now read a genome in only a few hours for under $1000. Short read sequencing is many times called "next generation" sequencing and is the method of choice these days.
The drawback is that there is no method for ensuring the exact length of the DNA segments and there is no way to keep them "in order." That is where I come in.
Step 2

The result from the sequencer is a data file composed of long strings of ACTG... etc. In order for the genome to be considered "sequenced" these strings have to be put in the correct order. The IEEE Spectrum article entitled The DNA Data Deluge by Michael Schatz and Ben Langmead sums up this step very well when they compare sequencer output to running the book "A Tale of Two Cities" through a paper shredder and then trying to reassemble the shreds. The process is made even more complicated by the fact that typically not just one strand of DNA is run through the shredder, rather for redundancy and error checking anywhere from 50 to 100 copies of the DNA strand are run at a time.
So to sum up, you've run 50 to 100 copies of "A Tale of Two Cities" through a paper shredder and gotten a bin full of paper slips with a random number of words on each, and you need to reassemble one copy of the book from that big mess.
The output from this step is a text file with a bunch of contiguous lengths of base pairs (called contigs) which can then be manipulated and annotated further.
Back Where We Started
Assembly and alignment both work with the Step 2 output.
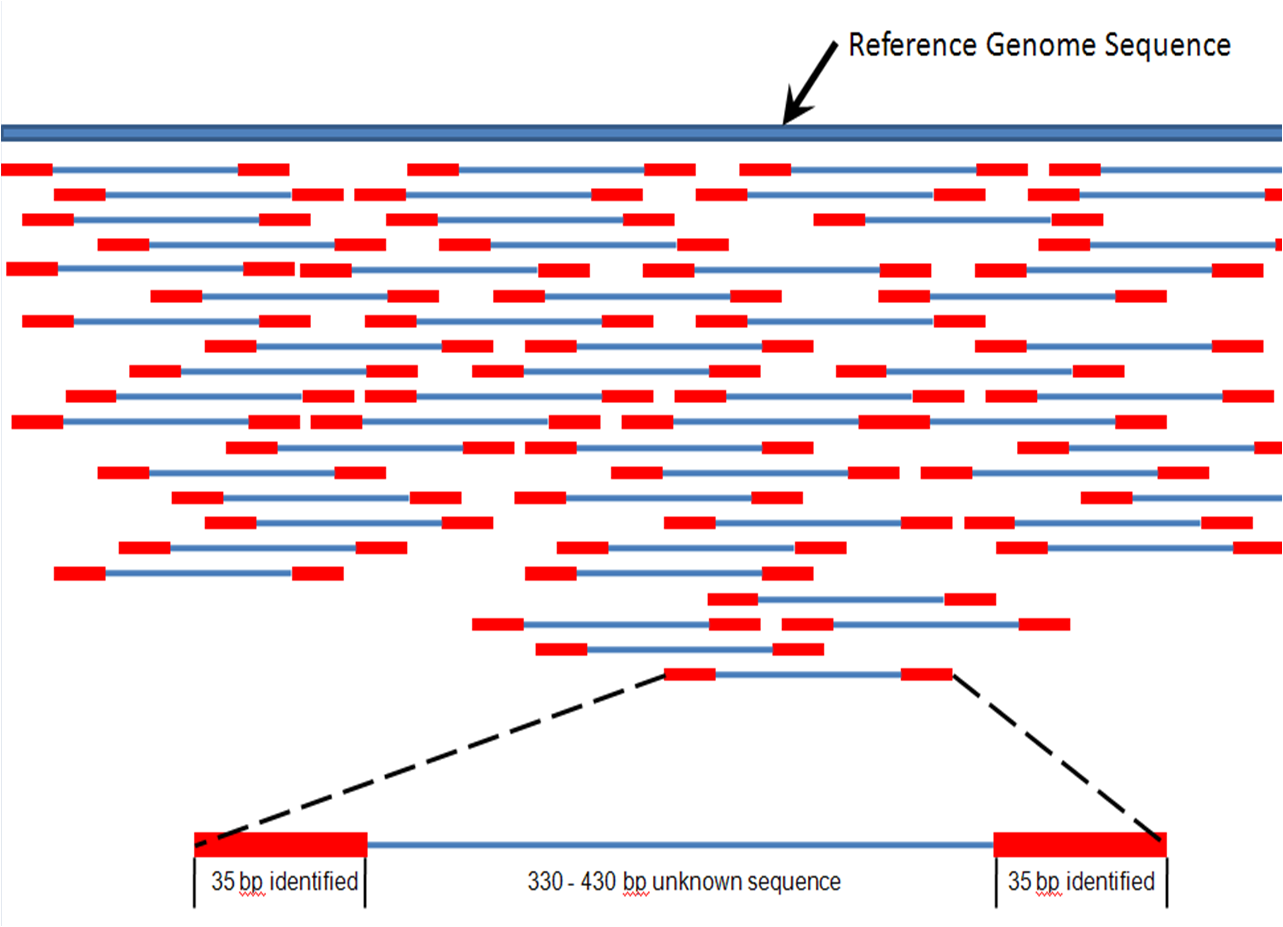
- Alignment takes the resultant paper slips and compares them, word by word, to a fully assembled version of "A Tale of Two Cities," aligning each slip to its' proper place. Aligning is by far the much easier process.
- Assembly, in contrast, attempts to order the slips without a reference. Many times you will also see the Latin phrase de novo, meaning "for the first time" or to "begin again," associated with assembly. An example would be a de novo assembly of different humans than the reference.
As I mentioned in the last article, we are having some trouble with de novo assembly of short read data of more complicated organisms like mammals. An MIT article from 2011, High-quality draft assemblies of mammalian genomes from massively parallel sequence data, states they developed software that can assemble a mouse genome in 3 weeks and a human genome in about 3 1/2 weeks.
Always listen to experts. They'll tell you what can't be done, and why.Then do it.-- Lazarus Long, from Robert A. Heinlein's "Time Enough For Love"
Possibly because I'm new to this whole "bioinformatics" thing or possibly because I'm just too stupid to listen to experts who say "it can't be done," I think I've found a loophole that has been overlooked. I'm still not ready to talk about the loophole - primarily because I don't want to listen to said "experts" until I have some data to back up my claims. Until that time, y'all are just going to have to wonder what I'm going to pull out of my... hat.

* Actually, another genetic structure called RNA substitutes uracil for thymine, but that's a slightly different story and not relevant to what I'm showing :).
References:
Schatz, M. C., & Langmead, B. (2013). The DNA data deluge. Spectrum, IEEE, 50(7), 28-33.
Gnerre, S., MacCallum, I., Przybylski, D., Ribeiro, F. J., Burton, J. N., Walker, B. J., ... & Jaffe, D. B. (2011). High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proceedings of the National Academy of Sciences, 108(4), 1513-1518.
This is actually the first explanation of DNA sequencing I've ever understood, Mike, thanks! Also, I wouldn't use the word "stupid" referring to your not listening to "experts". I think "stubborn" (and a "good" stubborn at that) would be a better choice.
ReplyDeleteGood on you also for playing things close to the chest as you are.
I look forward to reading more about your work.
Thanks!
Bill